LoadingMetadata
This tutorial covers the shell scripts that can be used to load the metadata with the graphical interface, with screenshots, and the rake task, to be run in the command line, if you are more comfortable in the terminal. The assumption is that expVIP is located in ~/expvip-web/
The first thing to do is to setup the available factors. The factor file is a text file, where each field is separated by tabs. A header is necesary on each column. The headers are the following:
- factor: The name of the factor to group. These must match those used in the metadata file (see below).
- order: Default display order in the graphical interface.
- name: The long name of the grouped factor. These must match those used in the metadata file (see below).
- short: Short name of the grouped factor. This is used in the graphical interface when many factors are displayed.
factor order name short
Age 1 7 days 7d
Age 2 seedling stage see
Age 3 14 days 14d
Age 4 three leaf stage 3_lea
Age 5 24 days 24d
Age 6 tillering stage till
Age 7 fifth leaf stage 5_lea
Age 8 1 cm spike 1_sp
Age 9 two nodes detectable 2_no
Age 10 flag leaf stage f_lea
Age 11 anthesis anth
Age 12 2 dpa 2dpa
Age 13 4 dpa 4dpa
High level age 1 seedling see
High level age 2 vegetative veg
High level age 3 reproductive repr
High level stress-disease 1 none none
High level stress-disease 2 disease dis
High level stress-disease 3 abiotic abio
High level stress-disease 4 transgenic trans
High level tissue 1 spike spike
High level tissue 2 grain grain
...
- Double click in
load_factors.shin the desktop
- When prompted, run execute in terminal
- By default, the script goes to
/media, which is the folder containing theshared foldersthat we have setup in the LoadingVM step.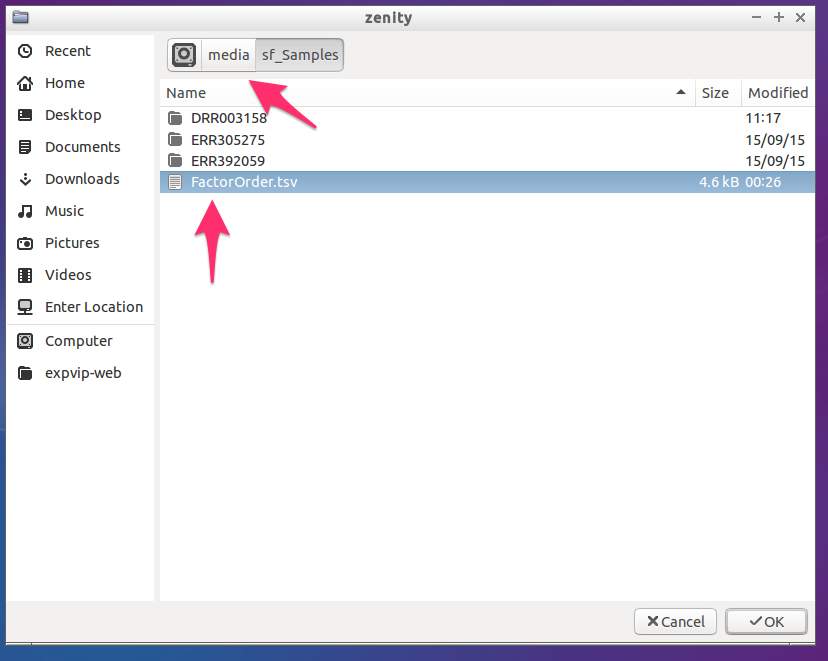
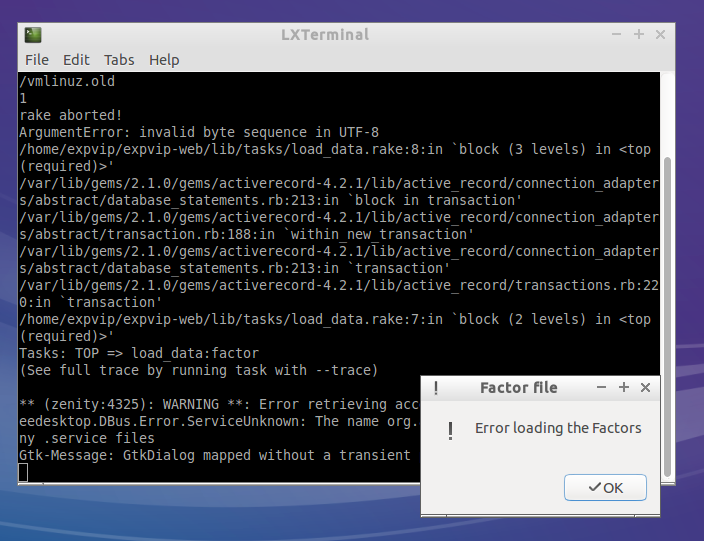
- If the factors are loaded correctly, a pop up window will notify about it
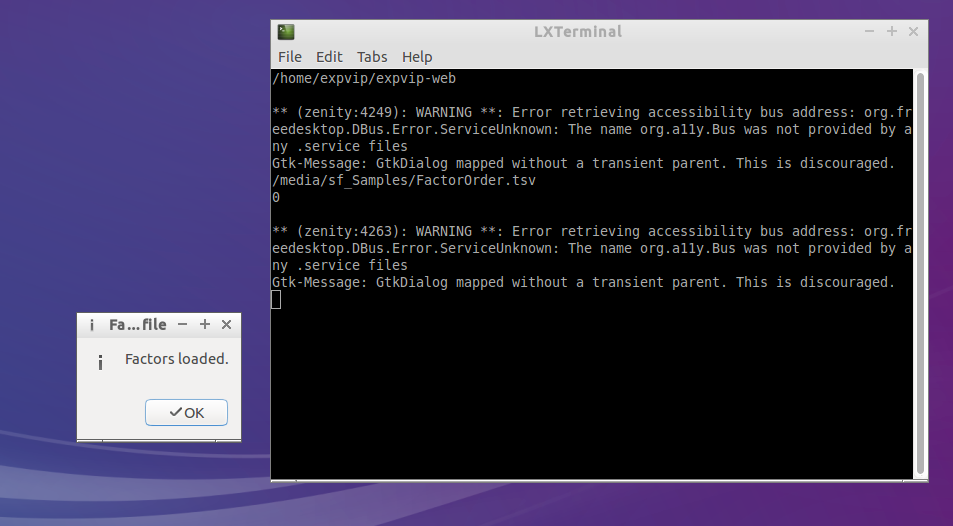
- If there was an error loading the factors, a message will notify about it. The error log may give a hint of what went wrong, but if you can't figure out send a screenshot of the terminal to the developers.
To load the factors, you can run directly the rake task from ~/expvip-web/.
rake load_data:factor[FILE_WITH_FACTORS];
The second step is to load the experiment metadata. Currently, a tab separated file is the input and it must contain the following columns with the header named exactly as stated:
- secondary_study_accession: The accession number for experiments carried as part of a single study. This is usually the high level BioProject or SRA number.
- run_accession: The accession of the individual run.
- scientific_name: of the species.
- experiment_title: A description for the individual RNA-seq sample.
- study_title: A description of the general study.
- Variety
- Tissue
- Age
- Stress-disease
- Manuscript: The DOI of the study.
- Group_for_averaging A description of the experiment. This must be the same all the replicates in the same study.
- Group_number_for_averaging: A short name for replicated experiments.
- Total reads: (optional)
- Mapped reads: (optional)
- High level variety: A higher level grouping to get summarized data of the factors.
- High level tissue
- High level age
- High level stress-disease
-
Variety,Tissue,Age,Stress-disease, and their correspondingHigh levelfactors must be exactly the same as in the columnsfactorandnamefrom thefactor file(see above). - The graphical interface will group samples based on these factors. Therefore these can be defined based on the user needs. For example the factor
High level tissuewill include tissue types such asgrain,roots,spikeandleaves/shoots. Within each of these tissue types, a more detailed description can be included under theTissueheading. For example:starchy endosperm,seed coat,transfer cells, etc. RNA-seq samples which share factor names in common will be displayed as groups in the visual interface. - If
Mapped readsandTotal readsare missing, you need to runkallistomapping from theraketask.
The process is similar to loading the factors. However, the metadata file is selected.
- Double click on the
load_metadata.shicon in the desktop
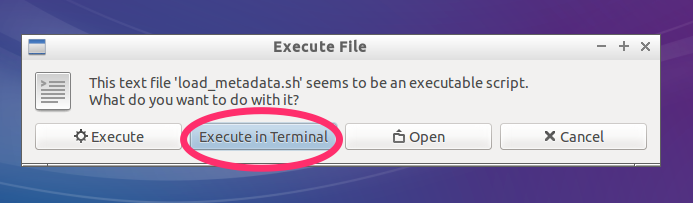
- Choose execute in terminal
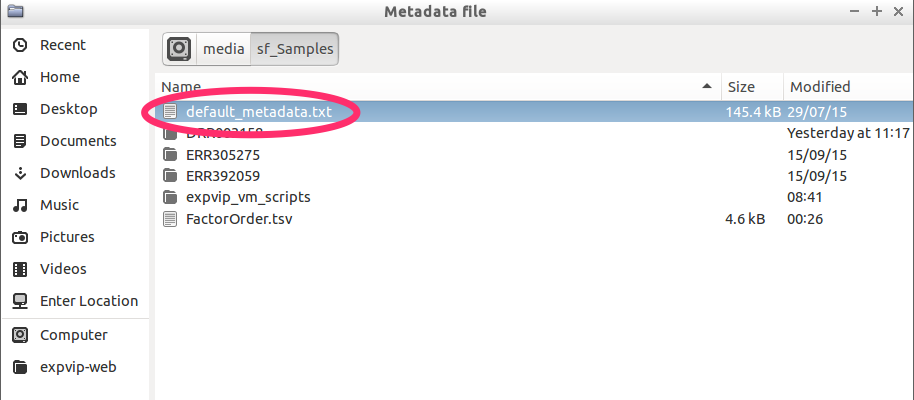
- Select the metadata file
rake load_data:metadata[FILE_WITH_THE_METADATA]Before loading the actual expression data or running kallisto, it is necessary to load the gene models. Currently, only the fasta file with the cdna from ensembl is supported. The fasta header should contain the following fields, besides the gene name (first string in the header).
- cdna
- chromosome or scaffold are converted to position
- gene
- transcript
- description a free text, in quotes. Any other field with quotes may fail in the load.
Besides the fasta file, it is necessary to give a name to the gene set. For this tutorial, the gene_set will be IWGSC2.26
>Traes_5BL_3FC5BA305.1 cdna:novel scaffold:IWGSC2:IWGSC_CSS_5BL_scaff_1082268:5:199:-1 gene:Traes_5BL_3FC5BA305 transcript:Traes_5BL_3FC5BA305.1
TGCTGCTGCTAGGCTTGAAGAGGTTGCTGGCAAGCTCCAGTCTGCTCGGCAGCTCATTCA
GAGGGGCTGTGAGGAGTGCCCCAAGAACGAGGATGTTTGGTTCGAGGCATGCCGGTTGGC
TAGCCCAGATGAGTCAAAGGCAGTAATTGCCAGGGGTGTGAAGGCAATTCCCAACTCTGT
GAAGCTGTGGCTGCA
>Traes_6BL_9BB648D51.1 cdna:novel scaffold:IWGSC2:IWGSC_CSS_6BL_scaff_430516:302:1741:-1 gene:Traes_6BL_9BB648D51 transcript:Traes_6BL_9BB648D51.1
TCCCTATCTGTTTCCTTGGCAGCTCCCTGATCCAATCGATCCATCAGGGCTCGACTAACT
TCTTCCAGCGCCTCTTCAGCGCGGGAGATCTACCAGCGTCGGCGGAGGGGCGTAGGTGCA
GGCGTGCAGCCCAAGTCCGCACCCGGCTCTAGGTTTCTGCTAATCTTCTTCCACCTGTGA
TACGCGCTCCGGGGCTAGGAGCACTCGTTGCCGGCTGCCTCGTGCTCGGAATGGCGGATG
- Double click on the
load_gene_setscript
- Select
Execute in Terminal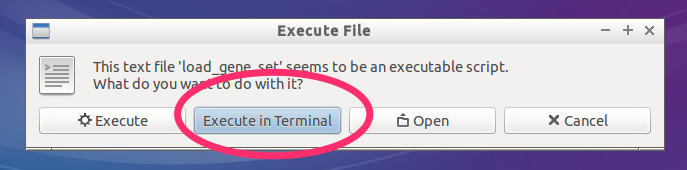
- Here you can name the gene set.
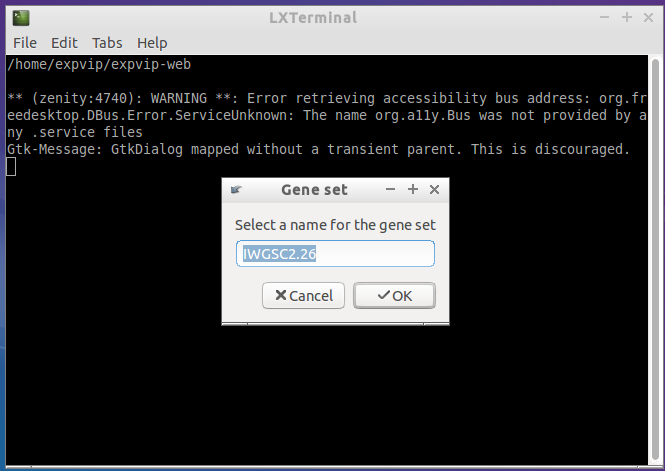
- And select the reference file. This may take a few minutes to load.
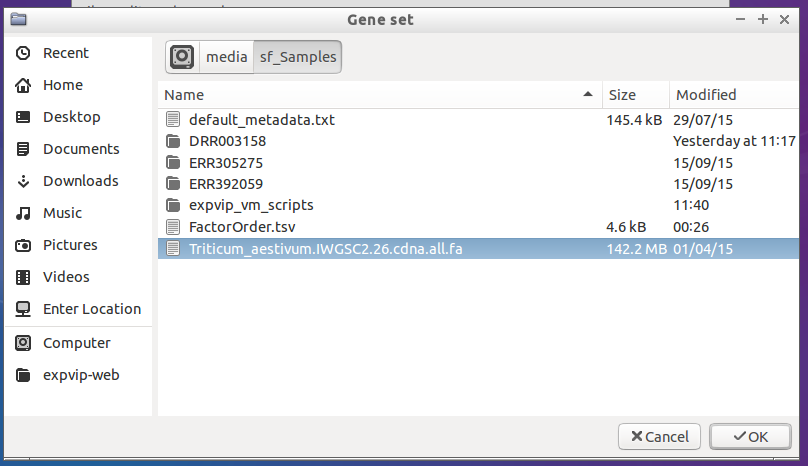
- Successfully loaded genes
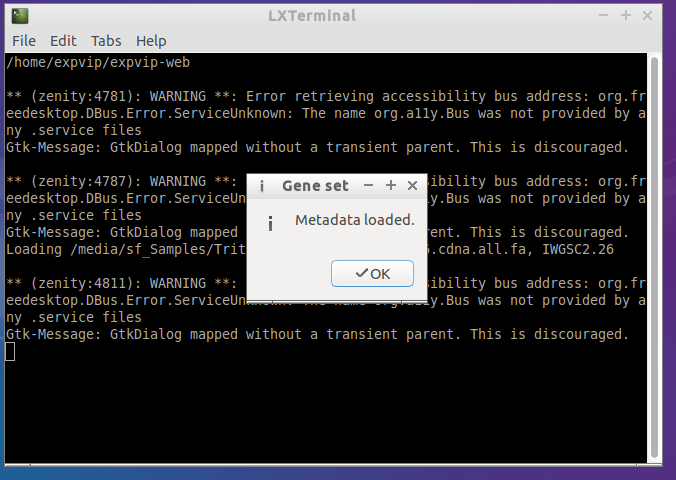
rake load_data:ensembl_genes[IWGSC2.26,/Triticum_aestivum.IWGSC2.26.cdna.all.fa]##Loading the homoeologues ## In order to show the homoeologues, a file with the homoeologies must be loaded. The file is tab separated with the following format:
Gene A B D Group Genome
Traes_5BS_0AFC3F795 Traes_5BS_0AFC3F795 Traes_5DS_C204EBAA9 5 B
Traes_5DS_C204EBAA9 Traes_5BS_0AFC3F795 Traes_5DS_C204EBAA9 5 D
Traes_7DL_82360D4EE1 Traes_7DL_82360D4EE1 7 D
Traes_2AL_1368BE0AD Traes_2AL_1368BE0AD Traes_2BL_CD459994C1 2 A
...
Note that the gene names are not the same as the transcript names, they correspond to the gene name.
The file can be genrated with ensembl compara, using the following query:
SELECT
homology_member.homology_id, cigar_line, perc_cov, perc_id, perc_pos,
gene_member.stable_id as genes,
gene_member.genome_db_id
FROM
homology_member
INNER JOIN homology USING (homology_id)
INNER JOIN method_link_species_set USING (method_link_species_set_id)
INNER JOIN gene_member USING (gene_member_id)
WHERE method_link_species_set.name="T.aes homoeologues";Then, to format the result of the query (saved as compara_homology.txt), you can use the provided script
ruby bin/homologyTable.rb compara_homolgy.txt homology.txt homology_counts.txtYou can get your homoeologies elsewhere, as long as you keep the file format.
At this point, the homoloeologues are called A,B and D. This is going to change on a future release to allow any chromosome group naming.
- Double click on the
load_homoeologues.shscript
- Select
Execute in Terminal
- Here you can name the gene set. It must be the same name you added for the gene reference.
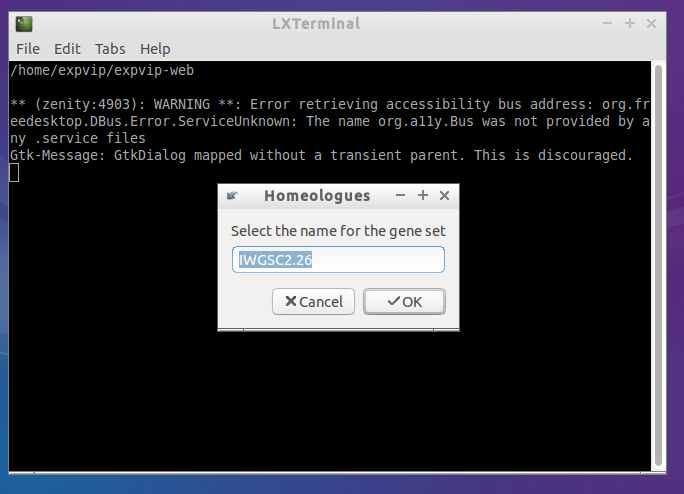
- And select the homoeologues file.
- Succesfully loaded. At the end of the log you can see which how many homologies where loaded.
rake load_data:homology[IWGSC2.26,/homology.txt]