This repository is for my research on plant mitochondrial genomes, which I call "mitogenome" in this README.md. Shortly and roughly speaking, mitogenomes of plants are different from those of animals or fungus. For example,
- The sizes are much larger.
- Genomes contain some repetitive regions, and
- (repeat-)mediated recombinations create divergent structures called "multipartite structure"s.
I'm trying to reconstruct these multipartite structures in the plant mitogenomes. In contrast to usual assembly workflows, I'm currently developping another way to assemble each structure; clustering.
Although it partly solves the problem of mitogenome reconstruction, it does depend on a 'good' reference genome. I'm working on this repository to adress this issue.
- Author: Bansho Masutani @ University of Tokyo
- Contact: [email protected]
The following list is the list of accessions/URLs to the data used in this research:
- Seven accessions of Arabidopsis thaliana via Sequence Read Archive
- One Ler accession from PacBio's official repository
- One Col-0 accession newly sequenced at the National Institute of Genetics in Japan.
All of these data were generated by Sequel Systems. For more details, e.g., the used chemistory, please visit the links.
The Gen Bank IDs of the reference sequences used in this research are BK010421.1, JF729100, and JF729102. BK010421.1 is the reference sequence I'm using generally.
WGS reads were filtered to enrich reads from mitochondrial genome. Specifically, we run the following command:bash ./script/filter_reads ${WGS} ${BK010421.1} > ${FILTERED}.
We re-assembled these datasets by JTK, and put the results here.
| Strain Name | Master circle (FASTA) | Master circle (GFA) | Alignments (BAM) |
|---|---|---|---|
| c24 | FASTA | GFA | BAM |
| pacbio | FASTA | GFA | BAM |
| sha | FASTA | GFA | BAM |
| col0 | FASTA | GFA | BAM |
| cvi | FASTA | GFA | BAM |
| eri | FASTA | GFA | BAM |
| kyo | FASTA | GFA | BAM |
| ler | FASTA | GFA | BAM |
| an1 | FASTA | GFA | BAM |
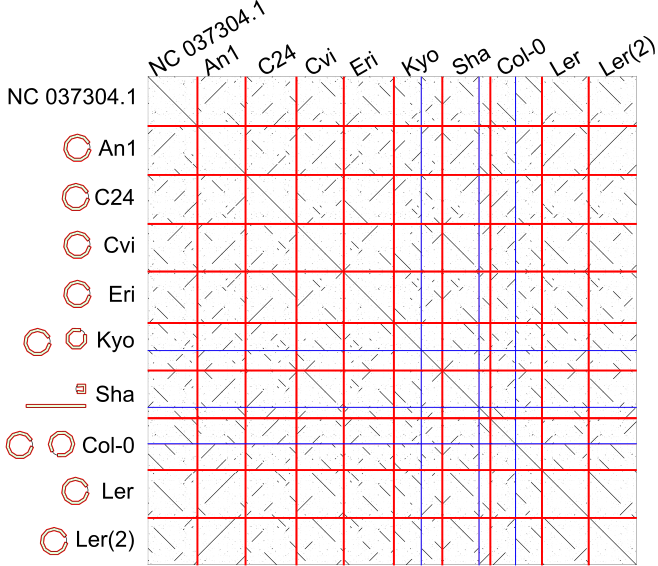
The dotplots between these strains as follows (or click here).
The red lines are boundaries between strains, and blue lines are boundaries of contigs.
The filtered reads, the outpot of PO-HMM, and supplementary plots are available at the following locations. "Circos," "linear," and "no-marge" represent a circos plot (like in the paper), a dot plot between assembled contig and the reference, and a circos plots without any "aggressive merging," respectively.
| Strain Name | Reads | Result | Circos | Linear | no_merge |
|---|---|---|---|---|---|
| pacbio | Reads | Result | Circos | Liner | NoMerge |
| ler | Reads | Result | Circos | Liner | NoMerge |
| col0_1106_exp2 | Reads | Result | Circos | Liner | NoMerge |
| cvi | Reads | Result | Circos | Liner | NoMerge |
| an1 | Reads | Result | Circos | Liner | NoMerge |
| c24 | Reads | Result | Circos | Liner | NoMerge |
| kyo | Reads | Result | Circos | Liner | NoMerge |
| sha | Reads | Result | Circos | Liner | NoMerge |
| eri | Reads | Result | Circos | Liner | NoMerge |
First, install the dependencies. Minimum requirements are
- BadRead:
git clone https://github.com/rrwick/Badread.git && pip3 install ./Badreadwould installBadReadto your local envirnment. - Flye
- Minimap2:
git clone https://github.com/lh3/minimap2 && cd minimap2 && makeandln -s ${PWD}/minimap2 ${HOME}/local/bin/minimap2or anywhere included in the $PATH variable. - Samtools
- HTSlib
- Last
- Rust
To obtain baseline result, please install
- WhatsHap:
pip3 install --user whatshap - isONclust:
pip install isONclust. - CARNAC-LR: Exec
git clone https://github.com/kamimrcht/CARNAC.git && cd CARNAC && make. Then, you can sim-link${PWD}/CARNAC-LR ${HOME}/local/bin/CARNAC-LRor anywhere included in the $PATH variable. Also, copyCARNAC/scripts/paf_to_CARNAC.pyinto./script/of this repository.
Then,
git clone --recursive https://github.com/ban-m/reconstruct_mito_genome.git
cd reconstruct_mito_genome
cargo check && cargo build && cargo build --release # Build binaries.
bash ./script/create_mock_genome.sh # create datasets under ${PWD}/data/synthetic_data/ by using `BadRead`
bash ./script/synthetic_dataset.sh
bash ./script/benchmark.sh # create results under ${PWD}/result/benchmark/
bash ./script/posterior_probability.job
bash ./script/baseline_mock_genomes.job # Invoke `isONclust`, `CARNAC-LR`, `WhatsHap`, and `Flye`would create all the result used in the paper. Note that each script would take long time to be done (from a few hours to a few days).
First, download the dataset and the reference genome.
bash ${PWD}/donwloads_files.shPlease type ll -ht ${PWD}/data/filtered_reads to confirm that all the dataset are properly donwloaded; md5sum values are as follows:
- e040ac3f8b047b7e84d38cb70faf8ecc an1.fasta
- 7b4b5f70ba29891a09a83786db5d8ad9 c24.fasta
- 00157130264b868bf35dd10828749a3d col0_1106_exp2.fasta
- 893394f4ef02fe4737fb26b7896ea98f cvi.fasta
- 26646b904a44bd564c84ccf5bcc6c7e9 eri.fasta
- 072427917074882e4ad9dbc4b03ad87d kyo.fasta
- 67b2f656d2eaa91126f36b6b111a665f ler.fasta
- 4a92ad3d6fe66732e2fcba4d76488b53 pacbio.fasta
- 69a0007d689ba6a797c0e975a0eb8092 sha.fasta
Then, run the pipeline on each dataset. Here, ${threads} is the number of threads to be used. We used ${threads}=24.
bash ./script/real_dataset.sh ${threads}It takes several days to complete on a 24 threads 2.0GHz computer.
This program depends on following libraries and binaries:
After install these requirements, do
git clone --recursive https://github.com/ban-m/reconstruct_mito_genome.git
cd reconstruct_mito_genome
cargo check && cargo build && cargo build --release
Then, target/release/mmmm --help would print help messages to stderr.
Basic usage would be
mmmm decompose --output ${OUTPUT} \
--reads ${READ} --contigs ${REFERENCE} \
--cluster_num ${MIN_CLUSTER} --threads ${CORES}
${READ} and ${REFERENCE} should be fasta files. ${MIN_CLUSTER} is the size of the cluster at each window, not the number of resulting cluster.
Disclaimer1: It requires a reference quality contigs for clutering. For those dataset without very good reference, I'm currently developing this repository.
Disclaimer2: As the clustering algorithm (PO-HMM) is very costly, this program is for very small genome(<1Mbp).
- Jiao, W., Schneeberger, K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat Commun 11, 989 (2020). https://doi.org/10.1038/s41467-020-14779-y