Tutorial to run the AMBER umbrella COM restraint code and derive the free energy of transfer profile for methanol through a DMPC membrane.

- AMBER16 or above (has COM umbrella restraint code)
- Python 2.7 (https://www.continuum.io/downloads)
- WHAM (http://membrane.urmc.rochester.edu/content/wham)
You can download this tutorial from github, the resulting zip file will not have any trajectories from the MD simulations described below due to file size limits. You may have to give the scripts executable permissions with chmod. Or runs scripts as:
- bash bash_script.sh
- python python_script.py
The scripts have been downloaded and tested on a linux machine. If you are using a Mac you may need to correct line endings within the scripts.
This tutorial uses the AMBER16 center-of-mass (COM) umbrella restraint code to determine the free energy of transfer profile for a methanol molecule through a DMPC membrane bilayer. The methanol molecule is first pulled from the center of the membrane out into the water phase. From the pulling step, we extract starting positions with methanol at 0, 2, 4, ..., 32 A from the membrane center. We run windows with methanol restrained at each of these positions. From the fluctuation in the z-position, we can construct the free energy profile using WHAM. Finally, we use the same information to derive the z-diffusion and z-resistance profiles and an estimate of the overall permeability coefficient.
There is a great deal of literature available on running z-restraint simulations, which I would encourage you to consult. A few examples include:
-
Bemporad et al: http://pubs.acs.org/doi/abs/10.1021/jp035260s
-
Orsi et al: http://pubs.acs.org/doi/abs/10.1021/jp903248s
-
Carpenter et al: http://dx.doi.org/10.1016/j.bpj.2014.06.024
-
Lee et al: http://pubs.acs.org/doi/full/10.1021/acs.jcim.6b00022
First we need a starting membrane bilayer PDB file and the coordinates and parameters for methanol.
Directory: ./parameters
You can follow the AMBER tutorial TUTORIAL A16: An Amber Lipid Force Field Tutorial: Lipid14 to obtain an equilibrated lipid bilayer (although one is included here).
Use antechamber to create AMBER parameters for methanol from the enclosed methanol PDB:
antechamber -i methanol.pdb -fi pdb -o MOH.mol2 -fo mol2 -c bcc -s 2
Then use tleap to convert the resulting MOH.mol2 into an AMBER library file (MOH.off):
tleap -f convert.leap
Next we place the methanol molecule at the center of the membrane (the z-distance between methanol and DMPC bilayer is zero).
Directory: ./placement
A study has shown that pulling from the middle of the membrane out allows faster convergence of PMFs rather than pulling from the water phase into the membrane:
- Filipe et al: http://pubs.acs.org/doi/abs/10.1021/jp501622d
You can use the python script to place the methanol molecule at z=0 from the DMPC bilayer center-of-mass:
./com_placement.py -i DMPC_72_relax.pdb -d methanol.pdb -z 0.0 > moh_center.pdb
Important note: The com_placement.py script defines the center-of-mass of the membrane using the PC N31 head group atoms in the lipids. It will not work if these are not present: if you wish to use other atoms, you'll have to update the code. Also, if your drug molecule contains unusual atom types, you may need to add the atomic mass of these atoms into the com_placement code.
We can now create AMBER prmtop and inpcrd files of the system. First, to set the periodic box dimensions correctly, we use the vmd_box_dims.sh script from the A16 Lipid tutorial:
./vmd_box_dims.sh -i DMPC_72_relax.pdb -s water
48.158000, 47.372001, 77.938003
Using the output we can build the system with build.leap file shown below:
source leaprc.protein.ff14SB
source leaprc.water.tip3p
source leaprc.gaff
source leaprc.lipid14
loadoff ../parameters/MOH.off
drug = loadpdb moh_center.pdb
bilayer = loadpdb DMPC_72_relax.pdb
mol = combine {drug bilayer}
set mol box {48.158 47.372 77.938}
saveamberparm mol DMPC_MOH.prmtop DMPC_MOH.inpcrd
quit
Now run tleap:
tleap -f build.leap
Now that we have our system constructed, we first equilibrate it then run a pulling step, which slowly moves the methanol molecule from z=0A out into the water phase (z=32A).
Directory: ./pulling
First, we need the distance restraint file. You can use the make_COM_file.py script to construct this. It contains the atom indices of the drug atoms (group 1 to be constrained) and the atom indices of the lipid N31 head group atoms (reference to constrain to). It also contains details of the harmonic restraint to apply and flags to turn on the umbrella COM method.
First we need a pdb of the system with atom indexing correct:
ambpdb -p DMPC_MOH.prmtop < DMPC_MOH.inpcrd > for_index.pdb
Now run the script:
./make_COM_file.py -i for_index.pdb -o ref_COM_file.RST
As an aside, we can also check that we have the correct atom indices directly from the prmtop using parmed. We need an input for parmed, see details_parmed.in:
parm DMPC_MOH.prmtop
printDetails *
quit
Run ParmEd:
parmed.py -i details_parmed.in > atom_list.out
You can check that in atom_list.out the N31 atom index values corresponds to those in ref_COM_file.RST
Please go through the AMBER manual so that you know what each line in the ref_COM_file.RST means. Important flags are:
rk2=2.5 restraint force constant
fxyz=0,0,1 turn on umbrella COM in z-direction only
outxyz=1 print position of restrained molecule in x,y,z dimensions
You may notice that ref_COM_file.RST has settings "DISTHERE" - we will copy this to a new file and add in the correct settings here.
cp ref_COM_file.RST COM_dist.RST
Change DISTHERE to 0.0 in COM_dist.RST.
We also need a file for the pulling step COM_pull.RST. This is similar to COM_dist.RST but specifies a starting positon of 0 and a final position of 32, the force constant for pulling is also reduced to 1.1.
We now have the .RST files for equilibration (methanol is held at z=0) and for the pulling (methanol is moved from z=0 to z=32 A).
The inputs and run script are provided. Please also examine the input files, importantly:
&wt type='DUMPFREQ', istep1=1000 / print position of restrained molecule every 1000 steps
&wt type='END', /
DISANG=COM_dist.RST details of the COM restraint
DUMPAVE=04_Equil_dist.RST file to write position of restrained molecule to
You can then run both the equilibration (heat, hold methanol at z=0 for 100ps) and the pulling (move methanol from z=0 to z=32 A over 32 ns of simulation) with the following bash script:
./run_pull.sh
Note that the pulling rate is implicitely set by the total pull distance (32A, as set in COM_pull.RST) and the total simulation time (32 ns, as set in 05_Pull.in). The pulling rate here is then 1A per ns.
You will have to modify GPU / AMBERHOME specific information, or make it suitable for your cluster. This took 12 hours on a single GPU. However you can skip to the next step without doing the actual run, as the starting windows we extract from the the resulting trajectory are provided.
You can check the pulling step has worked by plotting the z-position:
xmgrace 05_Pull_dist.dat

We can now extract windows with 2 A spacing along the z-axis and run windows from each.
All outputs from the simulation have been moved into "./md_output" (minus the trajectory files).
First we need to extract starting points for each window run from the pulling trajectory.
Directory: ./windows
Important: We must create an imaged trajectory to extract these windows from in which the bilayer center-of-mass, as defined by N31 head group atoms, is imaged to the origin (0,0,0). This means that when we extract the position of the methanol molecule, we also know that this is the separation between the bilayer COM and the methanol too.
Run cpptraj with image.trajin file:
trajin ../pulling/md_output/05_Pull_DMPC_MOH_reduce.nc
center mass origin :2-217@N31
image center origin familiar
vector c0 :1 :1 out c0.out
trajout bilayer_zero.nc netcdf
Note: the 05_Pull_DMPC_MOH_reduce.nc trajectory included here from the pulling step is much reduced from the original output to keep within file size limits - it contains only 33 frames allowing the window extraction for the purposes of this tutorial. If you run the full pulling step of 32 ns you should obtain a trajectory with 16000 frames.
Then:
cpptraj DMPC_MOH.prmtop < image.trajin
This should output two files: the imaged trajectory from which we extract the snapshots, and the c0.out file which contains the postion of the methanol at each frame of the trajectory. We also know that this corresponds to the separation between the methanol and bilayer center-of-mass.
We can extract the window starting points using extract_window.py:
./extract_window.py -i bilayer_zero.nc -p DMPC_MOH.prmtop -d c0.out -start 0 -end 32 -space 2
This will output frames with the methanol at 0, 2, 4, ..., 32 A from the bilayer center-of-mass. These restarts are provided.
Now we can run each window for 5 ns using the 06_Prod.in input and run_window_cuda.sh bash run script.
Note on windows with negative z-value
The COM code is set up to also restrain molecules at a negative position along the z-axis, however there are some subtleties that may catch you out. If your starting z-position is below zero, the molecule will be held at the negative of the r2 value in the .RST file (whether the r2 setting is positive OR negative) - i.e. treat r2 as an absolute value, if the starting z-position is negative, the molecule will be restrained at the negative of this absolute value. Furthermore, the Rcurr value printed in the output file is always an absolute value. You can check the actual separation between the bilayer and drug center-of-mass either in the dist.dat file printed out, or using cpptraj. You may need to do some testing to to run windows with negative z-position.
The upshot of this is that the pulling simulation may have gone in the negative direction. This is not an issue if you use a symmetric bilayer. You should extract windows as:
./extract_window.py -i bilayer_zero.nc -p DMPC_MOH.prmtop -d c0.out -start 0 -end -32 -space -2
For simplicity, flip the -2, -4, ..., -32 windows to be positive with cpptraj:
cat << EOF >trajin
trajin frame_-2.rst
rotate * x 180.0 y 0.0 z 0.0
trajout frame_2.rst restart
EOF
cpptraj *prmtop <trajin
Sampling: For the purposes of the tutorial, the windows are separated by 2 A, with each window being run to 5 ns and the z-position being written every 10th step (istep=10). However to obtain a better converged profile, I recommend 1 A spacing, 30 ns run time (at a minimum, ideally 100 ns or more) and istep=1. We will constrast results from 5 ns versus 30 ns run time later.
./run_window_cuda.sh
Important: You should run this using a COM_dist.RST file which is a copy of ref_COM_file.RST (i.e. it contains DISTHERE which gets substituted for the correct distance for each window).
If you have multiple GPUs you may want to split these steps into parallel runs, or run each over a CPU cluster. Each window takes roughly 2 hours, there are 17 windows. You can skip running the simulations if you prefer given that all outputs, with the exception of trajectories, are provided.
All outputs from the simulation have been moved into "./md_output" (minus the trajectory files). They are also zipped up, use the "unzip.sh" script to extract each dist directory.
Once the simulations are finished you can build the free energy profile with WHAM.
Directory: ./windows/md_output
The simulations should output a file called "06_Prod_dist.dat" (the name is given in the 06_Prod.in input). This has the format:
Frame# x: (x-coord) y: (y-coord) z: (z-coord) (total-coord)
Where each coord entry is the distance between the methanol and bilayer COM in each dimension. In this tutorial, we are only interested on the z-dimension. In the 06_Prod.in file, the setting to write to this file is istep1=10, so distances are written every 10th step (0.02 ps) meaning the resulting file can become large. If possible, it is better to write this data frequently (perhaps even every single step).
For WHAM and the next steps, we only need the z-dimension, so we can use an AWK script to extract this per window:
awk '{print $1,"",$7}' 06_Prod_dist.dat > prod_dist.dat
This is also possible for every window using the included bash script "fix_dist.sh".
Once you have prod_dist.dat files for every window (format: Frame# z-dist), we can run WHAM.
Directory: ./windows/wham_run
For wham input, you need a metadata file with the following information:
/path/to/file/distance.dat restraint-position force-constant
eg:
../md_output/dist_32.0/prod_dist.dat 32.0 5.0
../md_output/dist_30.0/prod_dist.dat 30.0 5.0
...etc...
You will notice that the force-constant value in metadata.dat is double that (5 kcal/mol/A^2) compared to that used in the simulations. This is due to differences in how restraints are defined in AMBER vs WHAM. Please see the WHAM documentation for more information.
You can prepare the metadata.dat file using the included script prepare_meta.sh - you may need to update this with the correct paths to your prod_dist.dat files.
Now run WHAM:
wham 0 32 320 0.00000001 303 0 metadata.dat out.pmf
Where the settings are:
wham (start) (end) (windows) (tolerance) (temperature) (padding) (input_metadata) (output)
You can then extract just the PMF curve and plot like so:
sed '1d' out.pmf | awk '{print $1,"",$2}' > plot_free_energy.dat
xmgrace plot_free_energy.dat
You should obtain a plot similar to this:

You will notice how shaky (unconverged) the profile is, below is the result for 30 ns windows with 1 A spacing and istep=1.

To examine how well the overlap is between each window, we can create a histogram of the drug z-position.
Directory: ./windows/wham_run/hist_plot
Use the run_hist.sh script to make a histogram from the prod_dist.dat files for each window (this calls the included generate_hist.py). You may need to check the file paths in run_hist.sh.
./run_hist.sh
You should get something like this:
xmgrace hist*dat

Again, you will notice the poor overlap of the windows. Below is the result for 30 ns.

We see that the overlap is suitable when using 30 ns windows, 1 A separation and 2.5 kcal/mol/A^2 force constant.
Important: The limits 0-> 32 A and bin number 160 are hard-coded into generate_hist.py, you will need to change this for windows at positions along the z-axis outside of these limits in your own simulations.
The final step computes first the diffusion along the z-axis, combines the result with the free energy profile data to obtain the resistance along the z-axis and finally integrates the resistance at each z-window to obtain an overall permeability coefficient estimate.
Directory: ./windows/wham_run/diffusion
For details on the position-dependent diffusion and resistance calculations please see the following publication from Gerhard Hummer: http://iopscience.iop.org/article/10.1088/1367-2630/7/1/034/meta
The following workflow is based on that by Lee et al (also linked at the top of this tutorial): http://pubs.acs.org/doi/full/10.1021/acs.jcim.6b00022
The position-dependent diffusion is calculated as:

Where

For each window, we must calculate the autocorrelation function of the Z-position from the restraint simulation then integrate the result up until it has decayed to zero. The position-dependent diffusion value D(Z) is then the variance var(Z) squared (i.e. the first value in the autocorrelation function squared) divided by the resulting integral. Here, we calculate the ACF and from it a D(Z) estimate using 1 ns periods. Given that we ran each window for 5 ns, we then have five estimates for D(Z) per window and can take an average over these.
The article from Lee et al has a thorough discussion on calculating position-dependent diffusion values (and inherent issues involved) so is advised reading.
If you ran 06_Prod.in for 5 ns, with istep1=10, then the final output distance file will have 250000 entries (i.e. a Z-position for every 0.02 ps). So we have 50000 samples per ns.
The script ACF_parse.cpp is adapted from the Rowley Lab (also included in the SI of the Lee paper): https://github.com/RowleyGroup/ACFCalculator
It has been adapted to work with outputs from the AMBER umbrella z-restraint COM code. To calculate the ACF from a 1 ns sample you can run it as follows:
Compile:
g++ ACF_parse.cpp -o ACF_calc.xRun for the z=32.0 window:
./ACF_calc.x -f ../../md_output/dist_32.0/prod_dist.dat -s 50000 -n 50000 -d 0.02 -o acf_plot.dat
Where:
- -f denotes the input file, which has two columns, the time (column 1) and the Z-position (column 2)
- -s is the number of samples (numSamples)
- -n is the number of samples over which to calculate the ACF (nCorr)
- -d is the timestep dt between samples in ps (here 0.02 ps)
- -o denotes the output file to which to write the ACF
- -c cut-off is an optional flag, which cuts off calculation of the integral of the ACF after the ACF has dropped to cut-off*variance (e.g. 0.01-0.05)
If you take a 1 ns sample from the z=32 A window and calculate the ACF and plot the resulting output file, you will see that it quickly decays to zero (within 5 ps), after which it oscillates around zero with a lot of noise. This noise affects the integral - instead, we only integrate up to the point that the ACF has decayed to 0.01 of its initial value using the -c flag:
./ACF_calc.x -f ../../md_output/dist_32.0/prod_dist.dat -s 50000 -n 50000 -d 0.02 -c 0.01 -o acf_plot.dat
You should obtain a value for the overall diffusion coefficient of about 3.26e-5 cm^2/s.
If you now do the same using a sample from z=0 A, you will see that although the ACF decays quickly, it bounces back up with a much more slowly decaying tail.

This means that our method of only integrating up until 0.01*variance is no longer acceptable. We instead need to integrate up until about 50 ps, given that the ACF decays roughly to zero by this time (50/0.02=2500, so we use 2500 samples):
./ACF_calc.x -f ../../md_output/dist_0.0/prod_dist.dat -s 50000 -n 2500 -d 0.02 -o acf_plot.dat
For methanol at least, the transition between using the cut-off method to using 50 ps integration window occurs at z=8 A.
You can use the following scripts to automate these calculations, please examine each so you know what they are doing and check that file paths are correct:
First:
./get_cut_int_diffusion.sh >> all_diffusion_values.out
Then:
./get_nCorr_diffusion.sh >> all_diffusion_values.out
If you change write frequency to istep=1, remember to set -d 0.002 and increase numSamples etc appropriately.
Now that we have the Z-dependent diffusion values D(Z), we can combine these with the value of the free energy at each Z-position to get the local resistance value R(Z) as:
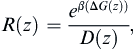
Once we have done that calculation we integrate over each R(z) value to get an effective resistance, the inverse of which is the permeability coefficient:
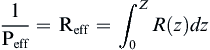
(The water layer is taken as z=0 for this integral).
I would urge you to do such calculations using a spreadsheet, so that you understand each step. A corresponding spreadsheet is enclosed.
A script to perform each step is also enclosed, called parse_fe_diff.py. This reads in the free energy profile, the diffusion profile and takes the z-limits plus step (i.e. 0->32 A, 2 A step) and the simulation temperature then calculates the resistance and does the integration.
Directory: ./windows/wham_run/overall_perm
./parse_fe_diff.py -fe ../plot.dat -diff ../diffusion/all_diffusion_values.out -start 0 -end 32 -space 2 -temp 303
This will output the free energy curve (free_energy_profile.parse.dat), the diffusion curve (diffusion_profile.parse.dat) and the resistance profile (resistance_profile.parse.dat) plus the overall permeability coefficient.
Note that the free_energy_profile.parse.dat has the same magnitude as that output from WHAM but is debased such that the free energy is zero in the water phase, whereas the WHAM output sets the global minimum to zero. For membrane PMFs, the standard way to report the free energy is with PMF at zero in the water phase (in the case, at z=32A).
Finally, we have only done the calculations for a single monolayer (water phase into the membrane center). If wish to get the values to move all the way through a symmetric membrane we can assume the values will be the same on the opposite side of the bilayer due to symmetry.
Directory: ./windows/wham_run/overall_perm/full_profile_perm
tac ../free_energy_profile.parse.dat | awk '{print $1*-1,"",$2}' > tmp
cat tmp ../free_energy_profile.parse.dat > full_fe.dat
rm tmp
tac ../../diffusion/all_diffusion_values.out | awk '{print $1*-1,"",$2}' > tmp
cat ../../diffusion/all_diffusion_values.out tmp > full_diffusion.dat
rm tmp
./parse_fe_diff.py -fe full_fe.dat -diff full_diffusion.dat -start -32 -end 32 -space 2 -temp 303
Will output the result for the full bilayer.
The resulting free energy profile:

The diffusion profile:

The resistance profile:

Corresponding profiles from 30 ns windows:
The resulting free energy profile:

The diffusion profile:

The resistance profile:

The values using 5 ns windows are:
- G(pen): 3.06 kcal/mol (free energy at the center z=0)
- P(eff): 0.296 cm/s
The values I obtain using 30 ns windows are:
- G(pen): 3.27 kcal/mol
- P(eff): 0.297 cm/s
Your values should be somewhere in this ballpark.
These compare favourably with those obtained by Orsi et al (also linked at the top of this tutorial): http://pubs.acs.org/doi/abs/10.1021/jp903248s
- G(pen): ~3.3 kcal/mol
- P(eff): 0.18 ± 0.2 cm/s
Convergence
Finally, there have been a number of articles addressing the issue of obtaining converged PMF profiles which you should take note of. Try to extend window simulation time and perform as many independent repeats from different initial coordinates and velocities to be sure your profiles are converged. Also, check from the histograms that your windows have suitable overlap.
A few papers you may want to read on the issues of convergence are linked below:
-
Paloncyova et al: http://pubs.acs.org/doi/abs/10.1021/ct2009208
-
Neale et al: http://pubs.acs.org/doi/abs/10.1021/ct200316w
Important: If you use the AMBER method and lipid force-field to generate PMFs for publication, please cite the relevant AMBER and Lipid14 (soon to be Lipid17) references:
- Dickson et al: http://pubs.acs.org/doi/abs/10.1021/ct4010307
Acknowledgements - thanks go to the following for adding in the COM umbrella restraint code, testing of the tutorial and general work on simulation of lipids in AMBER:
Ross Walker, Ian Gould, Charles Lin, Ben Madej, Aage Skjevik, Knut Teigen, Philip Morris, Mariarosaria Ferraro.